Welcome to DEcode!
The goal of this project is to enable you to utilize genomic big data in identifying regulatory mechanisms for differential expression (DE).
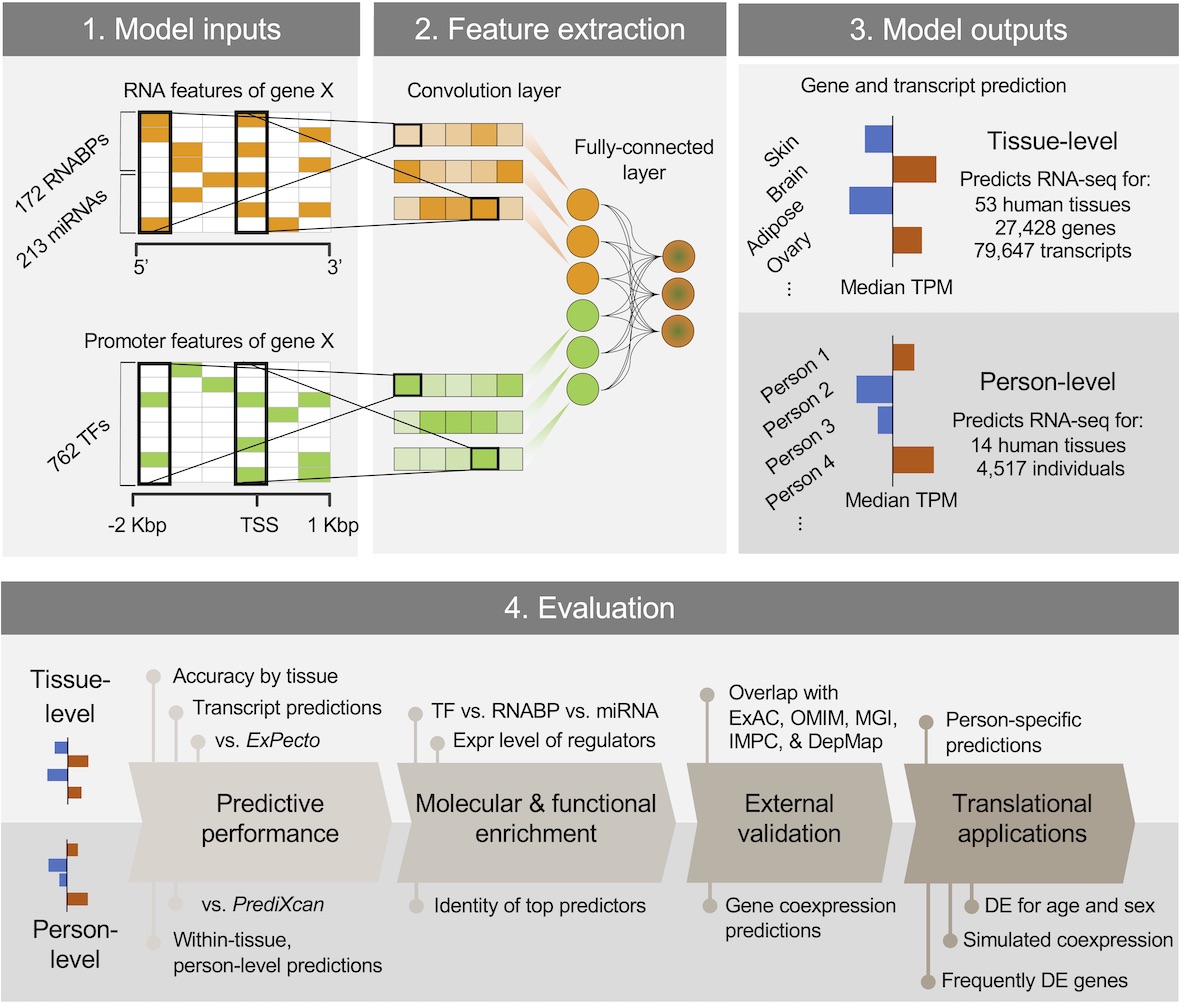
DEcode predicts inter-tissue variations and inter-person variations in gene expression levels from TF-promoter interactions, RNABP-mRNA interactions, and miRNA-mRNA interactions.
You can read more about this method in this paper (full text is available at https://rdcu.be/b5r3p) where we conducted a series of evaluation and applications by predicting transcript usage, drivers of aging DE, gene coexpression relationships on a genome-wide scale, and frequent DE in diverse conditions.
Run DEcode on Code Ocean
You can run DEcode on Code Ocean platform without setting up a computational environment. Our Code Ocean capsule provides reproducible workflows, all processed data, and pre-trained models for tissue- and person-specific transcriptomes and DEprior, at gene- or transcript level.
Prepare input features
Prerequisites
Before running the scripts to create custom input data, you must have the following python libraries installed on your system:
- pandas
- scipy
Also you need to install the following R packages:
- GenomicFeatures
- tidyverse
- data.table
- rtracklayer
- plyranges
- optparse
Usage
- First, download the example files by running the following commands: ```bash #GTF file from GTEXv7 (hg19) mkdir gtf wget https://storage.googleapis.com/gtex_analysis_v7/reference/gencode.v19.genes.v7.patched_contigs.gtf -P ./gtf/
#eCLIP-seq peaks from Encode (hg19) mkdir bed_rna wget https://www.encodeproject.org/files/ENCFF039BKT/@@download/ENCFF039BKT.bed.gz -P ./bed_rna/ wget https://www.encodeproject.org/files/ENCFF379UQU/@@download/ENCFF379UQU.bed.gz -P ./bed_rna/
#ChIP-seq peaks from Encode (hg19) mkdir bed_promoter wget https://www.encodeproject.org/files/ENCFF553GPK/@@download/ENCFF553GPK.bed.gz -P ./bed_promoter/ wget https://www.encodeproject.org/files/ENCFF549TYR/@@download/ENCFF549TYR.bed.gz -P ./bed_promoter/
This will create directories `gtf`, `bed_rna`, and `bed_promoter`, and download the GTF file, two eCLIP-seq bed files, and two ChIP-seq bed files into them.
Our input data for the gene-level model was constructed based on the gencode.v19.transcripts.patched_contigs.gtf file from the GTEXv7 dataset. This file contains only one representative transcript for each gene for the human genome (hg19).
If you use a different gene model, please make sure to filter GTF file and select a representative transcript for each gene before running the pipeline.
The input data for the transcript-level model was created based on `https://storage.googleapis.com/gtex_analysis_v7/reference/gencode.v19.transcripts.patched_contigs.gtf`. It is not necessary to filter the GTF file for the transcript-level model.
2. Convert genome coordinates to RNA coordinates using the following command:
```bash
Rscript functions/bed_to_RNA_coord.R -b ./bed_rna/ -n 100 -g gtf/gencode.v19.genes.v7.patched_contigs.gtf -t rna -o custom_RNA
Arguments
- bed_directory: Character string specifying the directory containing the bed files
- bin: Numeric value specifying the size of bins for the genomic features
- gtf_file: Character string specifying the path to the GTF file
- input_type: Character string specifying the experiment type of the bed files, i.e. “promoter” or “rna”
- output: Character string specifying the path and filename of the output file
This will convert bed files in the genome coordinates in the ./bed_rna/ directory to RNA coordinates using the gencode.v19.genes.v7.patched_contigs.gtf file and output as custom_RNA.txt.
If you want to map ChIP-seq peaks to promoters, use the -t option as promoter.
Rscript functions/bed_to_RNA_coord.R -b ./bed_promoter/ -n 100 -g gtf/gencode.v19.genes.v7.patched_contigs.gtf -t promoter -o custom_promoter
- To convert RNA-coordinate peaks to Pandas format, use the following command: ```bash python functions/to_sparse.py custom_RNA.txt python functions/to_sparse.py custom_promoter.txt
clean up
rm custom_RNA.txt rm custom_promoter.txt
This will convert the RNA-coordinate peaks in the `custom_RNA.txt` file and the `custom_promoter.txt` file to sparse Pandas DataFrames (`custom_RNA.pkl` and `custom_promoter.pkl`).
4. Place `custom_RNA.pkl`, `custom_RNA_gene_name.txt.gz`, and `custom_RNA_feature_name.txt.gz` in the directory where RNA features are located, for example: `./data/toy/RNA_features/`. Also place `custom_promoter.pkl`, `custom_promoter_gene_name.txt.gz`, and `custom_promoter_feature_name.txt.gz` in the directory for promoter features, for example: `./data/toy/Promoter_features/`.
5. Modify the code (Run_DEcode_toy.ipynb) as follows:
```python
mRNA_data_loc = "./data/toy/RNA_features/"
mRNA_annotation_data = ["POSTAR","TargetScan","custom_RNA"]
promoter_data_loc = "./data/toy/Promoter_features/"
promoter_annotation_data = ["GTRD","custom_promoter"]
This modification to the code will instruct it to utilize the custom_RNA.pkl and custom_promoter.pkl files as part of the RNA annotation data and promoter annotation data, respectively.
If you find DEcode useful in your work, please cite our manuscript.
Tasaki, S., Gaiteri, C., Mostafavi, S. & Wang, Y. Deep learning decodes the principles of differential gene expression. Nature Machine Intelligence (2020) [link to paper] (full text is available at https://rdcu.be/b5r3p)
Source databases for traning data.
- GTEx transcriptome data - GTEx portal
- Transcription factor binding peaks - GTRD
- RNA binding protein binding peaks - POSTAR2
- miRNA binding locations - TargetScan